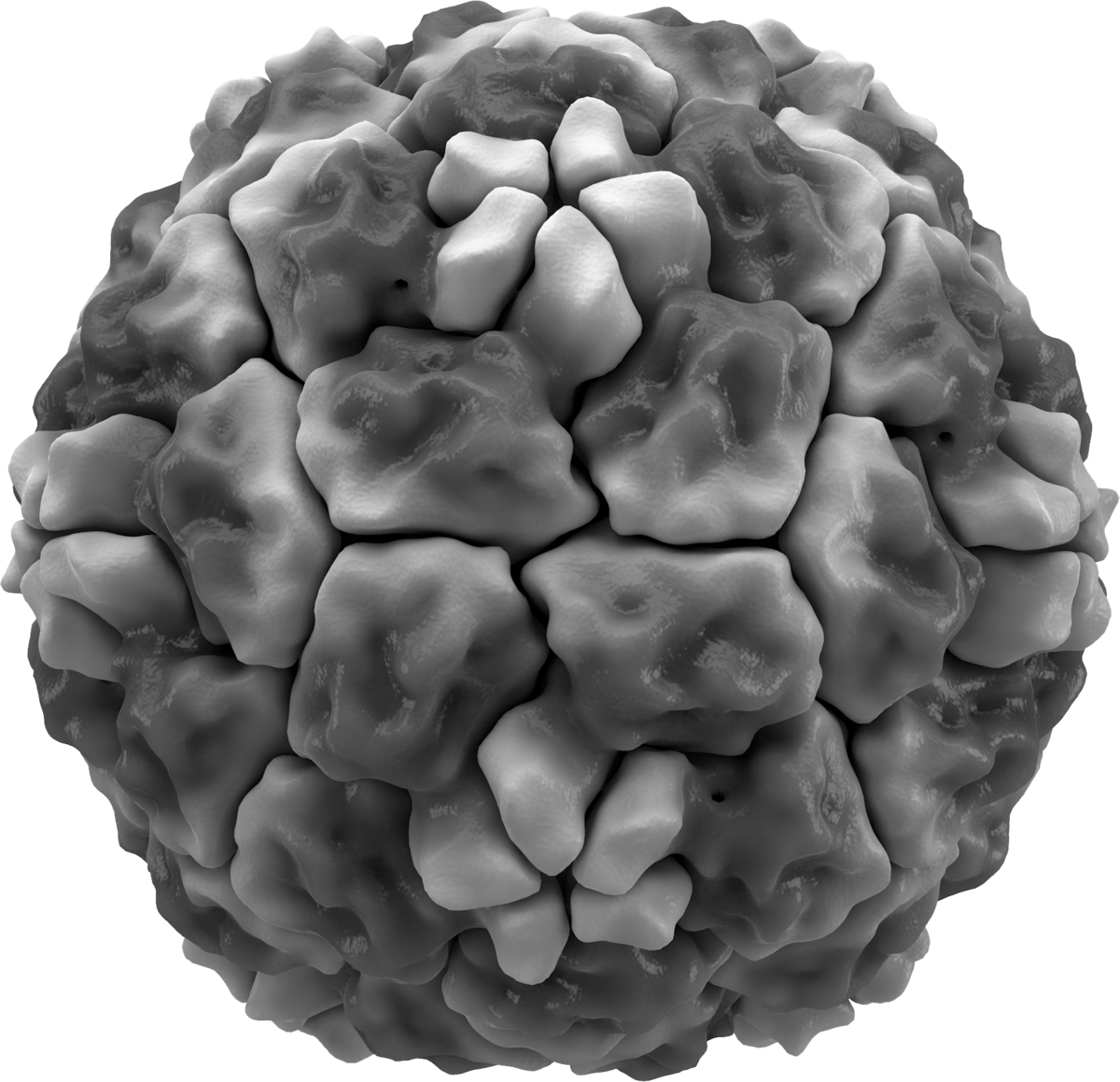
The common cold virus is part of the coronavirus (CoV) familiy with their characteristic spikes. All CoV spike proteins, that play an important role when a virus enters a host cell, are homotrimeric class I fusion proteins. These have three identical units of polypeptide in the so-called S1 domain that mediates receptor binding. The three units can be buried between their neighbors on the protein, called the closed state. Or they rotate and are sticking out, called the open state. In this state they can bind to a receptor but are also vulnerable to antibody attack. For SARS and MERS CoV for example, we know that the spikes can spontaneously switch between a closed or open configuration, balancing host cell attachment and immune evasion. However, most CoVs, notably the common cold CoV HKU1, appear to be tightly closed. How then, does it invade a host cell?
Modeling the spike protein
ICI researcher Joost Snijder at Utrecht University specializes in protein-glycan interactions. Recently, it was found that the HKU1 spike targets specific sialic acid receptors: α2,8-linked 9-O-acetylated disialosides which are glycan motifs typical of oligosialogangliosides such as GD3. Snijder says: "We knew from Geert-Jan Boons' work that HKU1-A binds to sialic acid, but we were not sure if this would be the receptor and where it would bind. That would give us crucial information on how this virus infects the cell."
Snijder and his team set out to investigate receptor binding of HKU1. "We cooperated with Daniel Hurdiss (Veterinary virology at UU) who is an expert on cryogenic electron microscopy of viral proteins. The challenge was producing sharp high quality EM-reconstructions to build atomic models, which has been the work of ICI postdoc Matti Pronker, and alining those with the results of molecular dynamics simulations." This resulted in the most detailed structure of the unbound HKU1-A spike protein so far.
Binding triggers opening
But the real surprise for Snijder came after incubating the HKU1 spike protein with the disialoside receptor analogue. They observed a remarkable conformational change in the spike protein after binding. They identified two more distinct conformations in addition to the closed state: a partially opened state with a single S1B domain rotated upwards and a fully opened state with three S1B domains up. These domains are located at the other end of the protein at a distance of 40 Å from the disialoside receptor. Snijder: "We did not anticipate this large conformational change so far away from the binding site. That is remarkable. This mechanism is fundamental to how this virus enters the cell."
A question mark in the publication remained the identity of the second receptor to which the opened S1B domains would bind. Demonstrating the importance of this research, within weeks other researchers had published the answer to that question: the receptor is TMPRSS2, a usual suspect in CoV research. "We would have investigated that ourselves, but this saves time," smiles Snijder. "The important thing is that our work has contributed to the knowledge on how to study this virus. Others can take advantage, which helps the field to progress."